
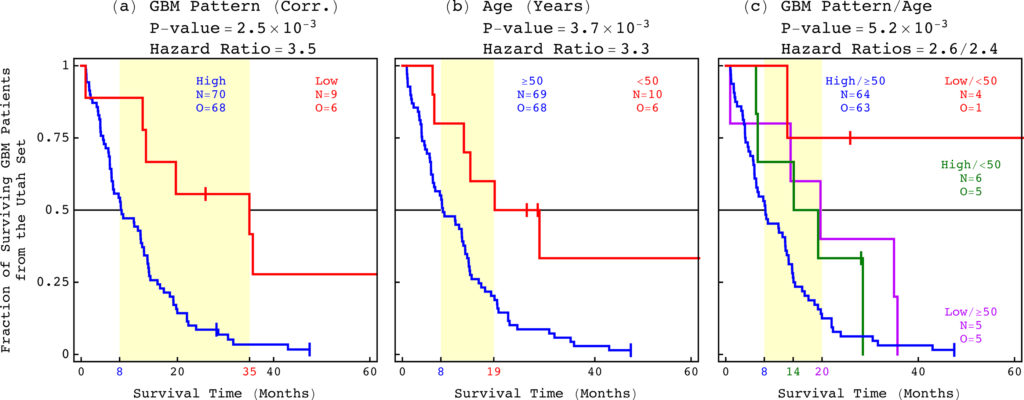
For the past 70 years, the best indicator of life expectancy for a patient with glioblastoma (GBM) — the most common and the most aggressive brain cancer — has simply been age at diagnosis. Now, an international team of scientists has experimentally validated a predictor that is not only more accurate but also more clinically relevant: a pattern of co-occurring changes in DNA abundance levels, or copy numbers, at hundreds of thousands of sites across the whole tumor genome.
Patients with the genome-wide pattern survive for a median of one year. However, patients without it survive three times as long, for a median of three years. The results came from a retrospective clinical trial that was published today in the journal Applied Physics Letters (APL) Bioengineering.
Having a predictor of a patient’s life expectancy can help inform medical decisions. The GBM pattern can, in principle, be used in this way today. For example, when a patient has magnetic resonance imaging results that are inconclusive such information can help doctors decide whether to perform an intervention.
“The information contained in this pattern, and other patterns that we can discover by using the same mathematical methods, can improve the standard of care of GBM and other diseases,” said the team leader Orly Alter, Ph.D., Utah Science, Technology, and Research (USTAR) associate professor of bioengineering and human genetics at the Scientific Computing and Imaging Institute and the Huntsman Cancer Institute at the University of Utah.
The international team of scientists has experimentally validated a predictor of a GBM patient’s life expectancy that is not only more accurate but also more clinically relevant than age at diagnosis, which has been the best indicator for the past 70 years.
Personalizing medicine
Having a predictor that encompasses the whole genome could also help drugs progress to regulatory approval. That is because the cumulative effect of all genes is thought to be involved in the drug response.
“We expect the GBM pattern, for example, to identify the patients who would benefit from a drug in prospective clinical trials,” said Sri Priya Ponnapalli, Ph.D., a senior research affiliate and an alumna of Alter’s lab, who is the first author of the paper.
“Even if the drug targets just one gene, a patient’s response to the drug would depend on the status of the whole genome and not just that one gene,” she added. The successful approval of any additional drug would be significant. In GBM, for example, only one drug, which does not cure GBM, has advanced from trials to standard of care over the last 40 years.
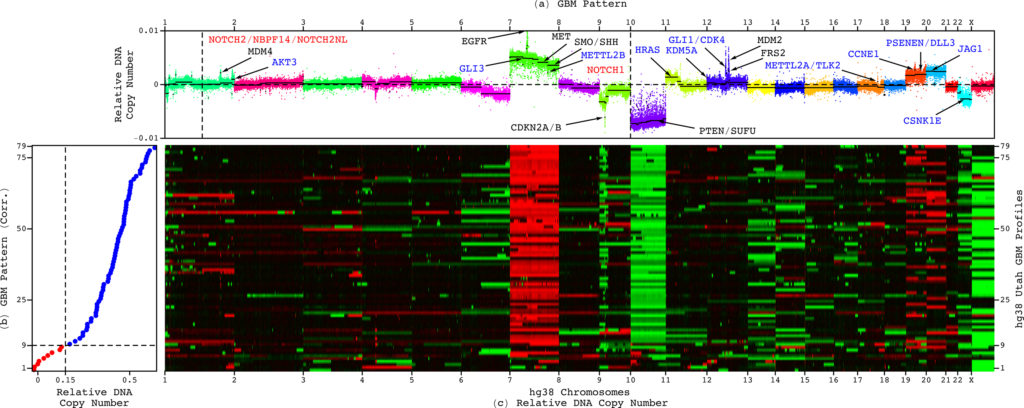
The GBM pattern not only predicts survival, but also includes most DNA copy-number alterations that were known and at least as many that were unrecognized in GBM prior to its discovery, including new druggable targets. This enables drug development efforts to directly focus on gene targets that have been shown to correlate with patient outcome.
Alter’s team were the first to successfully identify, and now to experimentally validate, a relationship between a GBM tumor’s DNA copy numbers and a patient’s life expectancy.
Finding relationships between patient outcomes and whole genomes
Alter’s team were the first to successfully identify, and now to experimentally validate, a relationship between a GBM tumor’s DNA copy numbers and a patient’s life expectancy. However, researchers have recognized this type of DNA alteration as a hallmark of cancer for more than a century and have observed them in GBM tumors for decades.
The team uncovered the relationship by developing and using new mathematical methods to compare and contrast tumor and normal whole genomes from the Cancer Genome Atlas (TCGA), a publicly open national database. Repeated previous attempts to associate a tumor’s DNA copy numbers with a GBM patient’s outcome failed, including previous studies of the same data from TCGA that used other methods.
This is the first predictor that encompasses the whole tumor genome.
The new mathematical methods have overcome three distinct challenges that others had not. First, they found consistent patterns across the hundreds of thousands of sites in whole genomes, which have three billion nucleotides. Second, they were able to do that across the tumor and the matching normal genomes simultaneously. Third, they were able to do so in small cohorts of patients, about 100, that are typical in clinical trials.
“Our mathematical methods succeeded because they use the complex structure of the datasets rather than simplifying or standardizing them as is commonly done,” said Alter. “This enables the separation of patterns which occur only in the tumor genomes from those that occur in the genomes of normal cells in the body and variations caused by experimental inconsistencies.”
These methods can be used with any kind of data, biological and otherwise. They can therefore be used to inform personalized approaches to various health conditions, including infectious disease.
###
About this study
To read the APL Bioengineering article go to:
https://doi.org/10.1063/1.5142559
In addition to Alter, Ponnapalli, and colleagues from the University of Utah, the study’s other authors include Jill S. Barnholtz-Sloan and colleagues from the Case Western Reserve University School of Medicine, Julie M. Gastier-Foster, Jay Bowen, and colleagues from Nationwide Children’s Hospital, and Huanming Yang and colleagues from the Beijing Genomics Institute and the China National GeneBank.
The study was supported by the National Cancer Institute’s Physical Sciences in Oncology Network, the USTAR Initiative, and the National Institutes of Health’s Office of the Director.
About University of Utah Health
University of Utah Health provides leading-edge and compassionate medicine for a referral area that encompasses 10% of the U.S., including Idaho, Wyoming, Montana and much of Nevada. A hub for health sciences research and education in the region, U of U Health has a $373 million research enterprise and trains the majority of Utah’s physicians, including more than 1,250 health care providers each year at its Schools of Medicine and Dentistry and Colleges of Nursing, Pharmacy and Health. With more than 20,000 employees, the system includes 12 community clinics and four hospitals. For ten straight years, U of U Health has ranked among the top 10 U.S. academic medical centers in the Vizient Quality and Accountability Study.